


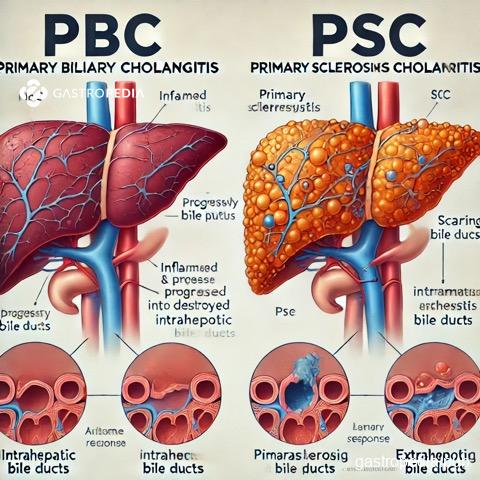
A colangite biliar primária (CBP) e a colangite esclerosante primária (CEP) são doenças hepáticas crônicas que afetam os ductos biliares, mas apresentam diferenças significativas em termos de patogênese, apresentação clínica e diagnóstico.
A CBP, anteriormente conhecida como cirrose biliar primária, é uma doença autoimune que afeta principalmente os ductos biliares intra-hepáticos. É mais comum em mulheres e frequentemente se apresenta com fadiga e prurido. O diagnóstico é geralmente confirmado pela presença de anticorpos anti-mitocondriais (AMA) em mais de 95% dos pacientes, juntamente com evidências bioquímicas de colestase, como elevação da fosfatase alcalina.[1][2]
Por outro lado, a CEP é caracterizada por inflamação e fibrose dos ductos biliares intra e extra-hepáticos, levando a estenoses multifocais. A CEP está frequentemente associada a doenças inflamatórias intestinais, especialmente a colite ulcerativa, e não possui um marcador sorológico específico como a CBP. O diagnóstico é geralmente feito por colangiografia por ressonância magnética (MRCP), que revela estenoses e dilatações características dos ductos biliares, conferindo uma aparência “em contas”.[3][4][5]
Em termos de complicações, a CEP está associada a um risco aumentado de colangiocarcinoma e outras malignidades, enquanto a CBP pode progredir para cirrose e insuficiência hepática se não tratada.[2][5] O tratamento para CBP inclui o uso de ácido ursodesoxicólico, que pode retardar a progressão da doença, enquanto para CEP, o transplante hepático é a única opção curativa, uma vez que não há tratamentos médicos eficazes disponíveis.[2][4]
Quais são os grupos de pacientes mais afetados por essas condições?
A CBP é uma doença hepática crônica que afeta predominantemente mulheres, com uma proporção de aproximadamente 9:1 em relação aos homens. A maioria dos casos é diagnosticada em mulheres de meia-idade, geralmente entre 40 e 60 anos.[1]
Por outro lado, a CEP é mais comum em homens, com cerca de 60% a 70% dos casos ocorrendo em pacientes do sexo masculino. A idade média de diagnóstico é geralmente entre 30 e 40 anos.[4] A CEP está fortemente associada à doença inflamatória intestinal (DII), especialmente à colite ulcerativa, que está presente em aproximadamente 70% dos pacientes com PSC.[3][6] A prevalência da PSC é maior em populações do norte da Europa e da América do Norte.[6]
Quais são os principais sintomas?
Na colangite biliar primária (CBP), os principais sintomas relatados pelos pacientes incluem fadiga e prurido. Esses sintomas são frequentemente debilitantes e podem impactar significativamente a qualidade de vida dos pacientes, embora não haja uma boa correlação entre a presença desses sintomas e o estágio da doença.[4]
Por outro lado, na colangite esclerosante primária (CEP), muitos pacientes são assintomáticos no momento do diagnóstico, sendo a condição frequentemente identificada por testes de função hepática anormais persistentes. Quando presentes, os sintomas mais comuns incluem fadiga, prurido e icterícia. Outros sintomas podem incluir desconforto abdominal no quadrante superior direito e perda de peso. A CEP também está associada a doenças inflamatórias intestinais, como a colite ulcerativa, o que pode complicar o quadro clínico com sintomas gastrointestinais adicionais.
Quais as opções de tratamento disponíveis para esses pacientes?
Para a colangite biliar primária (CBP), o tratamento de primeira linha é o ácido ursodesoxicólico (UDCA), que melhora os marcadores bioquímicos de colestase e está associado a uma melhor sobrevida livre de transplante hepático.[7][8] No entanto, cerca de um terço dos pacientes não responde adequadamente ao UDCA, necessitando de terapias adicionais. Nesses casos, o ácido obeticólico, um agonista do receptor farnesoide X, pode ser utilizado como terapia de segunda linha.[7][9] Além disso, agonistas do receptor ativado por proliferadores de peroxissoma (PPAR), como o seladelpar, estão sendo investigados e mostram resultados promissores.[9] Fibratos, como bezafibrato e fenofibrato, também são usados off-label para pacientes com resposta inadequada ao UDCA
Para a colangite esclerosante primária (CEP), atualmente não há terapias aprovadas que modifiquem a progressão da doença, além do transplante hepático, que é a única opção curativa.[7][10] O UDCA tem sido utilizado, mas sua eficácia em retardar a progressão da doença ou melhorar a sobrevida não foi comprovada Vários agentes estão em desenvolvimento, incluindo agonistas do receptor farnesoide X, como o ácido obeticólico, cilofexor e tropifexor, além de inibidores de ASTB/IBAP e análogos do fator de crescimento de fibroblastos (FGF)19.[11] A pesquisa continua a explorar novas terapias que possam abordar os mecanismos subjacentes da CEP, incluindo agentes que atuam na microbiota intestinal e nas vias de metabolismo dos ácidos biliares.[11]
| Característica | Colangite Biliar Primária (CBP) | Colangite Esclerosante Primária (CEP) |
|---|---|---|
Nome da Doença | Colangite Biliar Primária (CBP) | Colangite Esclerosante Primária (CEP) |
Patogênese | Doença autoimune, destruição progressiva dos ductos biliares intra-hepáticos | Inflamação e fibrose progressiva dos ductos biliares intra e extra-hepáticos |
Ductos Acometidos | Ductos biliares intra-hepáticos | Ductos biliares intra e extra-hepáticos |
Grupo mais afetado | Mulheres, 40-60 anos | Homens, 30-40 anos |
Associação com DII | Não associada | Forte associação com colite ulcerativa |
Sintomas Principais | Fadiga, prurido | Assintomática no início, fadiga, prurido, icterícia |
Diagnóstico | Anticorpos anti-mitocondriais (AMA) + bioquímica hepática | Colangiografia por ressonância magnética (MRCP) – imagem de ‘contas’ |
Complicações | Cirrose, insuficiência hepática | Colangiocarcinoma, cirrose, insuficiência hepática |
Tratamento | Ácido ursodesoxicólico (UDCA), ácido obeticólico, fibratos em casos refratários | Transplante hepático (única opção curativa), terapias experimentais em estudo |
Tabela 1: comparação entre colangite biliar primária e colangite esclerosante primária.
Referências
- Kwo PY, Cohen SM, Lim JK. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. The American Journal of Gastroenterology. 2017;112(1):18-35. doi:10.1038/ajg.2016.517.
- Yokoda RT, Carey EJ. Primary Biliary Cholangitis and Primary Sclerosing Cholangitis. The American Journal of Gastroenterology. 2019;114(10):1593-1605. doi:10.14309/ajg.0000000000000268.
- Chapman R, Fevery J, Kalloo A, et al. Diagnosis and Management of Primary Sclerosing Cholangitis. Hepatology (Baltimore, Md.). 2010;51(2):660-78. doi:10.1002/hep.23294.
- Lindor KD, Kowdley KV, Harrison ME. ACG Clinical Guideline: Primary Sclerosing Cholangitis. The American Journal of Gastroenterology. 2015;110(5):646-59; quiz 660. doi:10.1038/ajg.2015.112.
- Yimam KK, Bowlus CL. Diagnosis and Classification of Primary Sclerosing Cholangitis. Autoimmunity Reviews. 2014 Apr-May;13(4-5):445-50. doi:10.1016/j.autrev.2014.01.040.
- Weismüller TJ, Trivedi PJ, Bergquist A, et al. Patient Age, Sex, and Inflammatory Bowel Disease Phenotype Associate With Course of Primary Sclerosing Cholangitis. Gastroenterology. 2017;152(8):1975-1984.e8. doi:10.1053/j.gastro.2017.02.038.
- Bhushan S, Sohal A, Kowdley KV, Agaf FF. Primary Biliary Cholangitis and Primary Sclerosing Cholangitis Therapy Landscape. The American Journal of Gastroenterology. 2024;:00000434-990000000-01424. doi:10.14309/ajg.0000000000003174.
- Hasegawa S, Yoneda M, Kurita Y, et al. Cholestatic Liver Disease: Current Treatment Strategies and New Therapeutic Agents. Drugs. 2021;81(10):1181-1192. doi:10.1007/s40265-021-01545-7.
- Levy C, Manns M, Hirschfield G. New Treatment Paradigms in Primary Biliary Cholangitis. Clinical Gastroenterology and Hepatology : The Official Clinical Practice Journal of the American Gastroenterological Association. 2023;21(8):2076-2087. doi:10.1016/j.cgh.2023.02.005.
- Wheless WH, Russo MW. Treatment of Primary Sclerosing Cholangitis Including Transplantation. Clinics in Liver Disease. 2024;28(1):171-182. doi:10.1016/j.cld.2023.07.008.
- Fiorucci S, Urbani G, Di Giorgio C, Biagioli M, Distrutti E. Bile Acids-Based Therapies for Primary Sclerosing Cholangitis: Current Landscape and Future Developments. Cells. 2024;13(19):1650. doi:10.3390/cells13191650.
Como citar este artigo
Martins BC, Orso IRB. Qual a diferença entre colangite biliar primária e colangite esclerosante primária? Gastropedia; 2025 Vol 1. Disponível em: https://gastropedia.pub/pt/gastroenterologia/qual-a-diferenca-entre-colangite-biliar-primaria-e-colangite-esclerosante-primaria/

Professor Livre-Docente pela Faculdade de Medicina da Universidade de São Paulo
Médico Endoscopista do Instituto do Câncer do Estado de São Paulo (ICESP)
Médico Endoscopista do Hospital Alemão Oswaldo Cruz
Emerging Star pela WEO

Doutor em Ciências em Gastroenterologia pela USP
Especialista em Endoscopia Diagnóstica e Terapêutica da Gastroclínica Cascavel e do Hospital São Lucas FAG
Coordenador da Residência Médica em Cirurgia Geral e Professor de Gastroenterologia da Escola de Medicina da Faculdade Assis Gurgacz

{kind=link}