


A deficiência de lipase ácida lisossomal (LAL-D) é uma desordem crônica e progressiva do metabolismo dos lipídeos, agrupada em um grupo com cerca de 70 doenças de depósito lisossômico.
Apresenta um padrão de herança autossômica recessiva, causada por uma mutação no gene da lipase ácida lisossomal – LIPA, que resulta em ausência total ou deficiência significativa na atividade da enzima lipase ácida lisossomal (LAL). Mais de 120 mutações levando à perda de função da proteína já foram descritas no gene LIPA associadas à LAL-D, sendo a mais comum uma mutação em sítio de processamento de RNA mensageiro (splice junction mutation), E8SJM (c.894G>A).
Essa perda de função da proteína leva ao acúmulo de ésteres de colesterol (EC) e triglicérides (TG) dentro dos lisossomos, o que acarreta um desbalanço do metabolismo do colesterol, causando dislipidemia, aterosclerose precoce e disfunção orgânica.
A doença acomete uma ampla faixa etária, de recém-nascidos a adultos, com a maior parte dos casos diagnosticada antes dos 20 anos de idade. Homens e mulheres parecem ser igualmente afetados.
A prevalência exata da LAL-D permanece desconhecida, inicialmente, reportaram como sendo em 1 para cada 40.000 a 300.000 pessoas, dependendo da etnia e localização geográfica.
A suposição de que LAL-D é extremamente rara é baseada nos poucos casos publicados na literatura médica. Entretanto, a frequência de LAL-D nas diferentes populações não é conhecida e a doença, provavelmente, é subdiagnosticada.
Fisiopatologia
A LAL é responsável pela hidrólise de EC e TG, resultando em colesterol livre, ácido graxo livre e glicerol, que são liberados no citoplasma. A homeostase do colesterol é controlada, principalmente, pela concentração plasmática de colesterol livre, que influencia a atividade dos fatores de transcrição nuclear que regulam a síntese do colesterol e TG, expressão de receptores de LDL-c e efluxo do colesterol.
A diminuição ou ausência da atividade da LAL faz com que essa hidrólise de EC e TG fique reduzida, acarretando no acúmulo de colesterol livre e ácido graxo livre no interior dos lisossomos e redução dos seus níveis no citoplasma.
Apresentação Clínico-Laboratorial
LAL-D é uma doença heterogênea e se apresenta com sintomas, características e taxas de progressão que variam entre os indivíduos afetados. Essas diferenças se devem ao tipo de mutação no gene LIPA, o que leva à disparidade de níveis na atividade residual enzimática. O espectro da doença varia entre a forma grave e precoce, com alta mortalidade, que acomete crianças menores que um ano, conhecida por Doença de Wolman (DW), até a forma tardia, menos grave e de apresentação variável, que acomete crianças mais velhas e adultos, conhecida por Doença de Depósito de Éster de Colesterol (DDEC).
Na DDEC, a idade de apresentação varia desde crianças mais jovens até adultos, com a maior parte dos afetados entre 3 e 12 anos. A investigação diagnóstica, muitas vezes, é iniciada devido a quadro de dislipidemia em jovens, a aumento do volume abdominal causado pela hepatomegalia e à detecção ocasional de elevação de transaminases.
As manifestações clínicas mais comuns nesses pacientes são doença hepática e dislipidemia. Hepatomegalia foi descrita em 88-99% dos casos e esplenomegalia em 74-79% dos casos.
Esteatose hepática é comum e indivíduos afetados apresentam risco significativo de desenvolver fibrose e cirrose, com suas complicações, como hipertensão portal, ascite, encefalopatia hepática, varizes de esôfago e CHC. A partir do desenvolvimento dessas complicações, deve-se atentar para a necessidade de transplante hepático.
As manifestações extra-hepáticas mais comuns incluem: diarreia/esteatorreia, epigastralgia, náusea, anemia, colestase, atraso do crescimento e doenças cardiovasculares. Acúmulo de lípides no trato gastrointestinal é um achado frequente, relacionado, muitas vezes, com síndrome de má absorção.
Diagnóstico
O diagnóstico de LAL-D pode ser realizado por meio da redução da atividade da enzima lipase ácida lisossomal, mutação do gene LIPA e/ou biópsia hepática.
- Atividade da LAL
Atualmente, o exame mais utilizado para essa avaliação é o método de dried blood spot (DBS).

A atividade é medida utilizando-se um inibidor específico, chamado lalistat 2. Esse método resulta em uma boa separação na atividade da LAL em indivíduos controle normais, homozigotos e heterozigotos.

Mutação do gene LIPA
O sequenciamento completo das regiões de codificação do gene LIPA auxilia no diagnóstico e na caracterização de pacientes em investigação de LAL-D. Embora a maioria dos pacientes com LAL-D seja homozigota ou heterozigota composta para as mutações do gene LIPA, alguns pacientes possuem mutações intrônicas (sequência de nucleotídeos na qual um gene que é removido pelo RNA splicing durante a transcrição do produto final), que passam despercebidas pelo sequenciamento genético.

Biópsia hepática
O procedimento geralmente é realizado durante a suspeita diagnóstica de pacientes com LAL-D.
Macroscopicamente, o fígado de pacientes com LAL-D apresenta-se de coloração alaranjada ou amarelada.
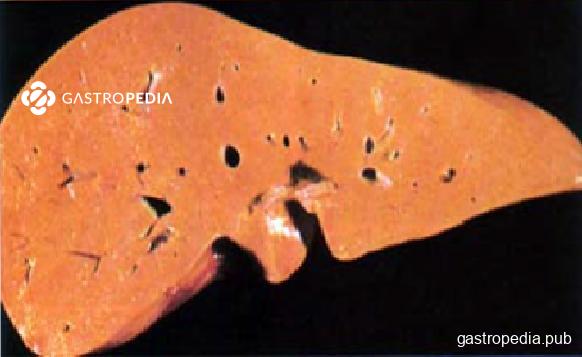
Fonte: Dincsoy et al., Am J Clin Pathol;
1984.
Na histologia, esteatose microvesicular ou mista geralmente está presente, porém esse achado, muitas vezes, não pode ser distinguido de outras causas de doença hepática gordurosa nem de uso de substâncias que podem levar a esse padrão histológico quando as lâminas são coradas com hematoxilina-eosina. Além disso, a luz polarizada pode ser utilizada para identificar cristais de colesterol em hepatócitos e células de Kupfer. Esses cristais birrefringentes podem ser examinados pela microscopia eletrônica e podem ser lisossomais ou citosólicos. A presença deles acredita-se ser patognomônica de LAL-D.
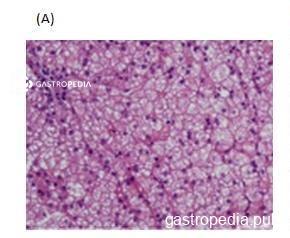
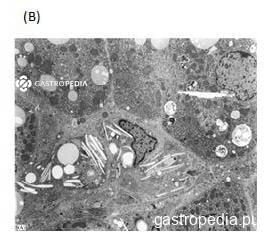
*Paciente do sexo masculino, 16 anos, com diagnóstico de CESD
Fonte: Laboratório de Patologia Clínica do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo
Alterações no trato gastrointestinal são observadas na mucosa e, menos frequentemente, na submucosa do intestino delgado. Macrófagos espumosos infiltrando a lâmina própria podem estar presentes, com distorção arquitetural e alteração na absorção e atividade enzimática dos enterócitos.

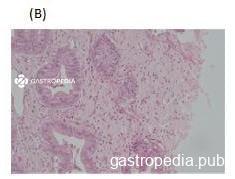
*Paciente do sexo feminino, 45 anos, com diagnóstico de DDEC
Fonte: Equipe de Endoscopia do Centro de Diagnóstico em Gastroenterologia (CDG) e Laboratório de Patologia Clínica do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo
Diagnóstico diferencial
Devido à semelhança com outras doenças cardiovasculares, hepáticas e metabólicas, o diagnóstico de LAL-D é desafiador. Sem a investigação apropriada, essas semelhanças podem levar ao diagnóstico errado e ao atraso do manejo apropriado.
Doenças que cursam com dislipidemia, Hiperlipidemia Combinada Familiar (FCH), Hipercolesterolemia Familiar Heterozigota (HeFH) e Hipercolesterolemia Poligênica estão entre os diagnósticos diferenciais.
Alterações hepáticas que se assemelham ao quadro de LAL-D podem incluir DHGNA, EHNA, doença hepática criptogênica e doenças de depósito lisossômico.
Tratamento
Antes da aprovação da terapia de reposição enzimática, com a sebelipase alfa (Kanuma®, Alexion Pharmaceuticals, Inc.), as opções terapêuticas para tratamento da LAL-D eram de suporte, incluindo agentes redutores da lipemia, dieta à base de baixa ingestão de gorduras, transplante de medula óssea e transplante hepático. Nenhum desses tratamentos, exceto a terapia de reposição enzimática (TRE), com a sebelipase alfa, que se demonstrou seguro e efetivo no tratamento da LAL-D.
Redução de fibrose hepática foi observada em 8 de 12 pacientes tratados com sebelipase alfa na semana 52 de tratamento (amostras obtidas pré-tratamento, nas semanas 20 e 52). Essa melhora histológica acompanhou a redução da gordura hepática, dos níveis de ALT e de LDL-c. Em três pacientes, não foi observada alteração do grau de fibrose e, em um paciente, mesmo com uso da TRE, houve progressão da fibrose hepática. A maior duração do tratamento tende a evidenciar melhor redução dos graus de fibrose.
Referências
- Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-43.
- Aslanidis C, Ries S, Fehringer P, Büchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33(1):85-93.
- Scott SA, Liu B, Nazarenko I, Martis S, Kozlitina J, Yang Y, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology. 2013;58(3):958-65.
- Carter A, Brackley SM, Gao J, Mann JP. The global prevalence and genetic spectrum of lysosomal acid lipase deficiency: a rare condition that mimics NAFLD. J Hepatol. 2019 Jan;70(1):142-50.
- Burton BK, Deegan PB, Enns GM, Guardamagna O, Horlen S, Hovingh GK, et al. Clinical features of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr. 2015;61(6):619-25.
- Muntoni S, Wiebusch H, Jansen-Rust M, Rust S, Seedorf U, Schulte H, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007;27(8):1866-68.
- Grabowski GA, Charnas L, Du H. Lysosomal acid lipase deficiencies : the wolman disease / cholesteryl ester storage disease spectrum. Lancet Gastroenterol Hepatol. 2017 Sep;2(9):670-9.
- Reiner Ž, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, et al. Lysosomal acid lipase deficiency – an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21-30.
- Wolman M, Sterk VV, Gatt S, Frenkel M. Primary familial xanthomatosis with involvement and calcification of the adrenals. Report of two more cases in siblings of a previously described infant. Pediatrics. 28:742-757.
- Jones SA, Rojas-caro S, Quinn AG, Friedman M, Marulkar S, Ezgu F, et al. Survival in infants treated with sebelipase Alfa for lysosomal acid lipase deficiency: an study. 2017;12(1):25.
- Drebber U, Andersen M, Kasper HU, Lohse P, Stolte M, Dienes HP. Severe chronic diarrhea and weight loss in cholesteryl ester storage disease: a case report. World J Gastroenterol. 2005;11(15):2364-6.
- Hůlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012;60(7):1107-13.
- Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413(15-16):1207-10.
- Su K, Donaldson E, Sharma R. Novel treatment options for lysosomal acid lipase deficiency: critical appraisal of sebelipase alfa. Appl Clin Genet. 2016; Volume 9:157-67.
- Burton BK, Balwani M, Feillet F, Baric I, Burrow A, Camarena C, et al. A Phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med. 2015;373(11):1010-20.
- Jones SA, Brassier A, Hughesc J, Plantazd D, Varae R, Breena C, et al. Effect of sebelipase alfa on survival and liver function in infants with rapidly progressive lysosomal acid lipase deficiency: 2-year follow-up data. Mol Genet Metab. 2016;117(2):S63.
Como citar este artigo

Gastroenterologia Clínica pela Faculdade de Medicina do ABC (FMABC)
Hepatologia pelo Hospital das Clinicas da Faculdade de Medicina de São Paulo (HCFMUSP)
Doutorado em Ciências em Gastroenterologia pelo HCFMUSP
Membro FBG e SBH
