


A Síndrome de Gilbert (SG) é uma desordem hepática do metabolismo das bilirrubinas com redução na glicuronidação da bilirrubina e consequente hiperbilirrubinemia indireta (não conjugada).
É uma condição comum (3-10% da população), com redução na atividade da UGT1A1 em 25-40%. As mutações ocorrem na sequência da região promotora (TATA box) do gene UGT1A1, a qual tem a função de controlar os níveis da proteína normal produzida. Desta forma, na SG, a proteína produzida é estruturalmente normal, porém em menor quantidade.
| Gene UGT1A1 Promove a produção da enzima bilirrubina-UGT, responsável pela conjugação de bilirrubina. Logo, mutações na UGT1A1 geram a produção de uma proteína anormal, com perda completa ou níveis menores de atividade da bilirrubina-UGT. |
Apresentação clínico-laboratorial
Clinicamente, os pacientes costumam ser assintomáticos e identificar elevações nos níveis de bilirrubinas totais com predomínio de bilirrubina indireta (<4-5mg/dL), de forma incidental, ou podem apresentar quadros intermitentes de icterícia, em especial, desencadeados por gatilhos como exercício físico intenso, baixa ingestão calórica/jejum, período menstrual, desidratação e infecções.
Laboratorialmente, não há elevação de enzimas hepáticas ou alterações nos demais exames de função hepática (tempo de protrombina e albumina), além de não haver indícios de hemólise ou doença estrutural fígado.
Saiba mais sobre alteração de enzimas hepáticas nesse post:
Diagnóstico diferencial
Distúrbios na captação hepática, armazenamento, conjugação e excreção podem ocasionar hiperbilirrubinemia. Dentre as causas hereditárias de hiperbilirrubinemia indireta, com exames normais de função hepática e sem alteração da histologia hepática, além da SG, faz-se o diagnóstico diferencial com:
- Síndrome de Crigler-Najar tipo I: condição muito rara com herança autossômica recessiva que se manifesta logo após o nascimento. Pela ausência da atividade UGT1A1 hepática, ocorre icterícia grave (20-45mg/dL ou mais) e risco de dano neurológico e óbito por kernicterus (encefalopatia bilirrubínica) nos primeiros dias após o nascimento. O tratamento precoce para a redução dos níveis de bilirrubina indireta no sangue é a fototerapia, devendo-se considerar a realização de transplante hepático como única terapia curativa.
- Síndrome de Crigler-Najar tipo II: condição rara com herança autossômica recessiva. Há atividade UGT1A1 hepática de 10% ou menos, com icterícia crônica (6-20mg/dL) e evolução potencialmente benigna. O tratamento com fenobarbital propicia a redução de cerca de 25-30% dos níveis de bilirrubina indireta pela indução da atividade da UGT1A1 residual.
Investigação diagnóstica
Identificada a hiperbilirrubinemia indireta, recomenda-se anamnese e exame físico detalhados, dosagem sérica de enzimas hepáticas (TGO, TGP, fosfatase alcalina e GGT) e função hepática (tempo de protrombina e albumina).
Caso haja alterações nesta primeira etapa de avaliação, direciona-se a investigação para a avaliação de hepatopatias, sendo prudente complementar com exame de imagem/ultrassonografia de abdome superior e demais exames laboratoriais específicos.
Se não forem identificadas alterações na primeira etapa de avaliação, é mandatório descartar hemólise com a dosagem de DHL, haptoglobina e reticulócitos.
Em adolescentes ou adultos, na ausência de hemólise e níveis de bilirrubina indireta <5mg/dL, presume-se o diagnóstico de síndrome de Gilbert. A confirmação é feita pelo teste genético para detectar mutações no gene UGT1A1/TATA box.
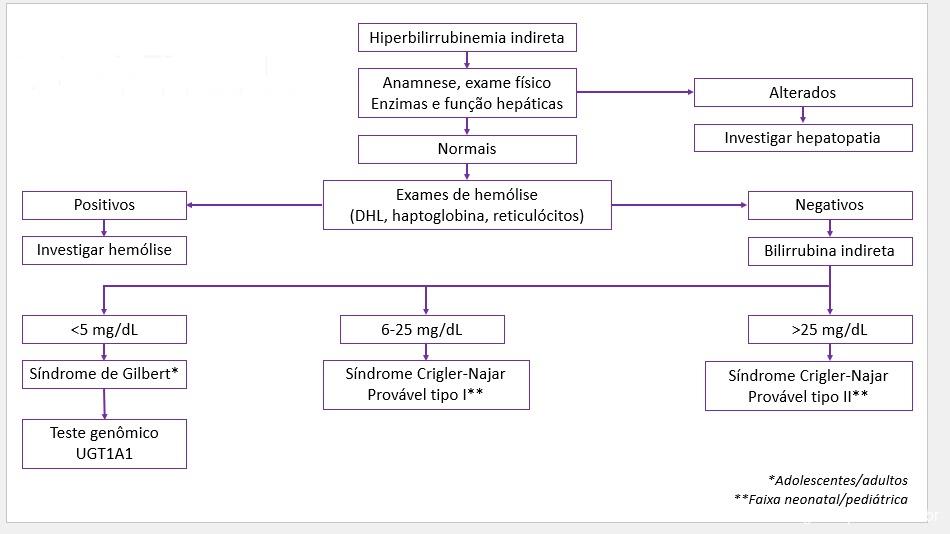
Figura 1. Fluxograma de investigação de hiperbulirrubinemia indireta.
Diagnóstico Genético
Diante da possibilidade de reações adversas a algumas drogas metabolizadas pelo UGT1A1, a exemplo do irinotecano e atazanavir, recomenda-se considerar a confirmação da SG pela pesquisa da mutação UGT1A1 pelo método de PCR em tempo real (Imagem 1).
Quando em homozigose, não há necessidade de rastreamento adicional, entretanto, se o paciente possuir apenas um alelo da mutação UGT1A1 ou ambos os alelos forem normais, deve-se pesquisar as mutações G71R e Y486D, as quais também se associam com a SG.

Imagem 1. Resultado do teste genético para Síndrome de Gilbert com homozigose do alelo 28 no gene UGT1A1.
Tratamento
Por ser uma condição benigna, o tratamento é conservador apenas com observação. O prognóstico dos pacientes com SG é excelente e não exige tratamento específico.
Referências
- Thoguluva Chandrasekar V, Faust TW, John S. Gilbert Syndrome. 2023 Feb 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 29262099.
- Singh A, Koritala T, Jialal I. Unconjugated Hyperbilirubinemia. 2023 Feb 20. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 31747203.
- King D, Armstrong MJ. Overview of Gilbert’s syndrome. Drug Ther Bull. 2019 Feb;57(2):27-31. doi: 10.1136/dtb.2018.000028. PMID: 30709860.
- Wagner KH, Shiels RG, Lang CA, Seyed Khoei N, Bulmer AC. Diagnostic criteria and contributors to Gilbert’s syndrome. Crit Rev Clin Lab Sci. 2018 Mar;55(2):129-139. doi: 10.1080/10408363.2018.1428526. Epub 2018 Feb 1. PMID: 29390925.
- Rodrigues C, Vieira E, Santos R, de Carvalho J, Santos-Silva A, Costa E, Bronze-da-Rocha E. Impact of UGT1A1 gene variants on total bilirubin levels in Gilbert syndrome patients and in healthy subjects. Blood Cells Mol Dis. 2012 Mar 15;48(3):166-72. doi: 10.1016/j.bcmd.2012.01.004. Epub 2012 Feb 9. PMID: 22325916.
Como citar este artigo
Oti KST, Síndrome de Gilbert: o que precisamos saber? Gastropedia 2023, vol. 2. Disponível em:
https://gastropedia.pub/pt/gastroenterologia/figado/sindrome-de-gilbert-o-que-precisamos-saber/
Gastroenterologia Clínica pelo Hospital das Clínicas da FMRP-USP. Hepatologia pelo Hospital das Clínicas da USP HCFMUSP. Doutorado em Ciências em Gastroenterologia HCFMUSP. Aperfeiçoamento de Elastografia Hepática Transitória HCFMUSP. Médica colaboradora do Ambulatório de NASH HCFMUSP. Membro FBG e SBH

{kind=link}