Procinéticos: Mecanismo de Ação, Indicações e Segurança
Os procinéticos são uma classe de fármacos utilizados no manejo de distúrbios da motilidade gastrointestinal. Seu mecanismo de ação baseia-se na estimulação das contrações do trato digestivo, favorecendo o esvaziamento gástrico e o trânsito intestinal. No Brasil, os principais representantes dessa classe incluem metoclopramida, domperidona, bromoprida, prucaloprida, neostigmina e eritromicina.
Este post resume o Expert Review da European Society of Neurogastroenterology and Motility em conjunto com The American Neurogastroenterology and Motility Society, que explora suas características farmacológicas, indicações clínicas e segurança.
1. Metoclopramida
A metoclopramida atua como antagonista dos receptores dopaminérgicos D2 e agonista parcial dos receptores serotoninérgicos 5-HT4, aumentando a liberação de acetilcolina no trato gastrointestinal. Esse mecanismo resulta no aumento da motilidade esofágica e gástrica.
Indicações:
- Gastroparesia diabética;
- Doença do refluxo gastroesofágico (DRGE);
- Náuseas e vômitos associados a cirurgias ou quimioterapia.
Efeitos adversos: O uso prolongado da metoclopramida está associado a um risco aumentado de efeitos adversos neurológicos, incluindo discinesia tardia irreversível, parkinsonismo, acatisia e distonia aguda. Esses efeitos extrapiramidais ocorrem devido à sua capacidade de atravessar a barreira hematoencefálica e antagonizar os receptores dopaminérgicos centrais. Além disso, pode causar sedação e sintomas autonômicos, como hipotensão ortostática. Devido a esses riscos, recomenda-se que seu uso contínuo não ultrapasse 12 semanas, conforme diretrizes da FDA.
2. Domperidona
A domperidona é um antagonista D2 que, ao contrário da metoclopramida, não atravessa a barreira hematoencefálica, reduzindo os riscos de efeitos extrapiramidais.
Indicações:
- Dispepsia funcional;
- DRGE;
- Gastroparesia leve a moderada.
Efeitos adversos: A domperidona pode prolongar o intervalo QT e aumentar o risco de arritmias ventriculares, especialmente em pacientes idosos ou com doenças cardiovasculares preexistentes. O risco cardiovascular é dose-dependente e pode ser agravado pelo uso concomitante de outros fármacos que prolongam o intervalo QT, como alguns antibióticos macrolídeos e antidepressivos tricíclicos. Recomenda-se monitoramento cuidadoso em pacientes de alto risco.
3. Bromoprida
A bromoprida compartilha o mesmo mecanismo de ação da metoclopramida, sendo um antagonista D2 com propriedades serotoninérgicas moderadas.
Indicações:
- Náuseas e vômitos de diversas etiologias;
- Gastroparesia leve;
- DRGE.
Efeitos adversos: Entre os efeitos colaterais mais comuns estão sintomas extrapiramidais, sedação e fadiga, o que pode limitar seu uso crônico.
4. Prucaloprida
A prucaloprida é um agonista altamente seletivo dos receptores 5-HT4, estimulando a liberação de acetilcolina no trato gastrointestinal e promovendo um aumento na motilidade intestinal, com maior impacto na motilidade colônica.
Indicações:
- Constipação crônica idiopática resistente ao tratamento com laxantes.
Efeitos adversos: Pode causar cefaleia, diarreia e dor abdominal. Seu perfil de segurança cardiovascular é favorável, sem associação relevante com prolongamento do intervalo QT.
5. Neostigmina
A neostigmina é um inibidor da acetilcolinesterase, aumentando os níveis de acetilcolina na junção neuromuscular e promovendo contrações no trato gastrointestinal.
Indicações:
- Pseudo-obstrução colônica aguda (síndrome de Ogilvie);
- Distúrbios de motilidade intestinal pós-operatórios.
Efeitos adversos: Os efeitos colaterais incluem bradicardia, cólicas abdominais, sudorese excessiva e hipersalivação. O monitoramento cardíaco é recomendado durante sua administração.
6. Eritromicina
Além de sua função antibiótica, a eritromicina atua como um agonista dos receptores de motilina, estimulando contrações gástricas semelhantes às do complexo motor migratório.
Indicações:
- Gastroparesia grave, especialmente em pacientes diabéticos;
- Esvaziamento gástrico antes de procedimentos endoscópicos;
- Pseudo-obstrução intestinal.
Efeitos adversos: Seu uso prolongado pode induzir taquifilaxia, além de interferir na microbiota intestinal e aumentar o risco de resistência bacteriana. Também está associada a prolongamento do intervalo QT em alguns pacientes.
Doses recomendadas:
| Metoclopramida: 10 mg via oral, intramuscular ou intravenosa, até 3 vezes ao dia (máximo de 30 mg/dia).
Domperidona: 10 mg via oral, até 3 vezes ao dia (máximo de 30 mg/dia). Bromoprida: 10 mg via oral ou intramuscular, até 3 vezes ao dia (máximo de 30 mg/dia). Prucaloprida: 2 mg via oral, uma vez ao dia (1 mg para idosos ou pacientes com insuficiência renal grave). Neostigmina: 0,5–2 mg intravenoso, administrado lentamente (monitoramento cardíaco recomendado devido ao risco de bradicardia). Eritromicina: 250–500 mg via oral, a cada 8 horas; ou 3 mg/kg intravenoso a cada 8 horas em casos de gastroparesia severa. |
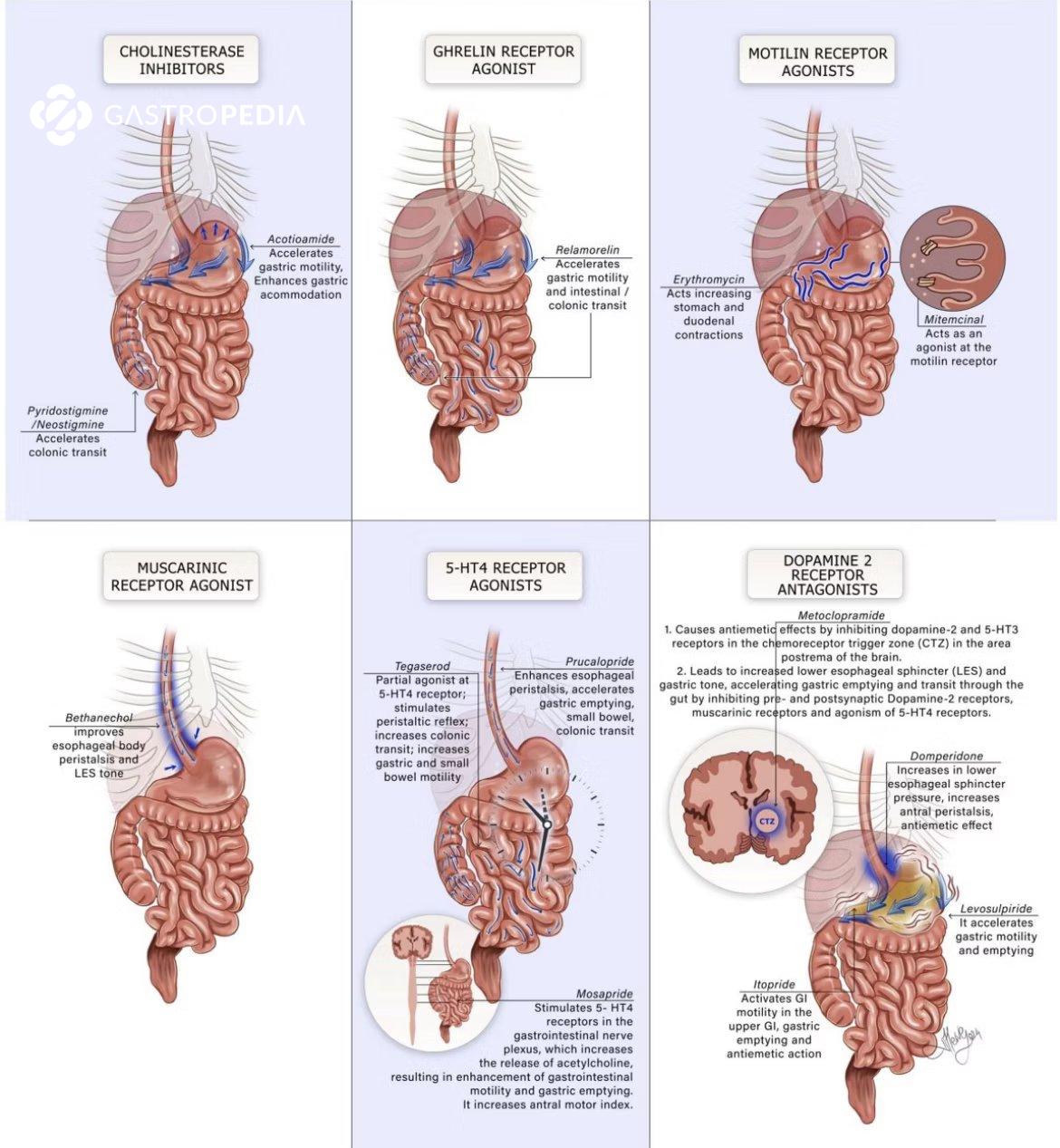
Ref: Bor S, et al. Neurogastroenterol Motil. 2024.
Considerações Finais
Os procinéticos têm um papel fundamental no tratamento de distúrbios da motilidade gastrointestinal, mas seu uso deve ser individualizado conforme o perfil clínico do paciente. Enquanto a metoclopramida, domperidona e bromoprida são indicadas para gastroparesia e DRGE, a prucaloprida é uma opção mais segura para constipação crônica idiopática. A neostigmina e a eritromicina são reservadas para casos graves, como pseudo-obstrução intestinal e gastroparesia severa. A decisão terapêutica deve equilibrar eficácia e segurança, evitando o uso prolongado quando possível.
Referências
- Bor S, Kalkan İH, Savarino E, Rao S, Tack J, Pasricha J, Cangemi D, Schol J, Karunaratne T, Ghisa M, Ahuja NK, Lacy B. Prokinetics-safety and efficacy: The European Society of Neurogastroenterology and Motility/The American Neurogastroenterology and Motility Society expert review.
Neurogastroenterol Motil. 2024 May;36(5):e14774. doi: 10.1111/nmo.14774. Epub 2024 Mar 10. PMID: 38462678.
Como citar este artigo
Martins BC. Procinéticos: Mecanismo de Ação, Indicações e Segurança Gastropedia 2025; Vol 1. Disponível em: https://gastropedia.pub/pt/gastroenterologia/procineticos-mecanismo-de-acao-indicacoes-e-seguranca/