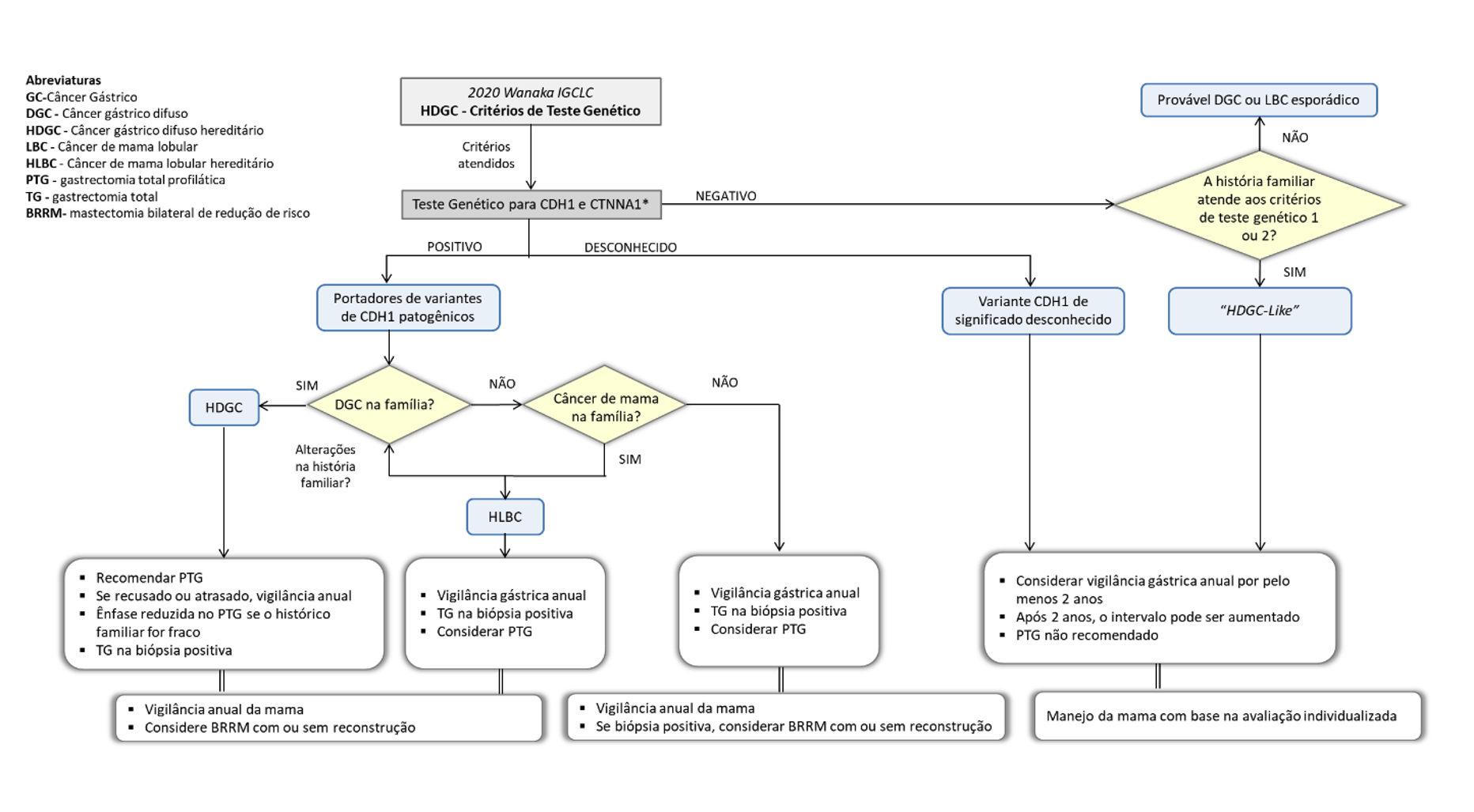
Como identificar e manejar os pacientes com suspeita de coledocolitíase?
Dez por cento dos americanos são portadores de colelitíase e apresentam sintomas relacionados aos cálculos biliares. No Brasil estudos retrospectivos mostram que a incidência de cálculos na vesícula biliar pode chegar a até 9,3%. Entre os pacientes com colelitíase, dez a vinte por cento podem apresentar coledocolitíase concomitante.
O advento da CPRE transformou o tratamento dos cálculos do colédoco de uma operação de grande porte em um procedimento minimamente invasivo. Porém, este procedimento apresenta uma incidência não desprezível de efeitos adversos como pancreatite, sangramento e perfuração. Devido a isso é importante selecionar de forma correta os pacientes que serão submetidos à CPRE para evitar a realização de exames desnecessários.
O guidelines da ESGE recomenda que os pacientes com colelitíase sintomática em programação de colecistectomia devem realizar a dosagem de enzimas hepáticas e ultrassom de abdome. Estes exames servem como triagem para avaliar o risco de coledocolitíase concomitante e determinar os pacientes que se beneficiariam de uma investigação adicional.
Avaliação do Risco de Coledocolitíase
A ASGE em 2010 publicou um guidelines contento uma lista de preditores para avaliar o risco de coledocolitíase.
Cálculos no colédoco visualizados na ultrassonografia, colangite e bilirrubina maior do que 4 mg/dl eram considerados preditores muito fortes. Dilatação das vias biliares e bilirrubina entre 1,8 e 4 mg/dl considerados com preditores fortes. Alteração de enzimas hepáticas, idade maior do que 55 anos e pancreatite biliar prévia preditores moderados.
Baseado nestes critérios os pacientes eram divididos em risco alto, intermediário e baixo risco. Para os de alto risco a recomendação era ir direto para a CPRE. Os de risco intermediário deveriam investigar melhor através da realização de colangiorressonância ou ecoendoscoscopia antes da decisão terapêutica. Os de baixo risco poderiam ir para colecistectomia direto.
Estudos posteriores utilizando estes critérios demonstraram que a sua acurácia era baixa, variando de 70 a 80%, com até 30% de procedimentos desnecessários.
Devido a isso, tanto a associação Americana (ASGE) quanto a europeia (ESGE) revisaram estes preditores em seus guidelines mais recentes.
Os novos critérios estão resumidos na tabela abaixo:
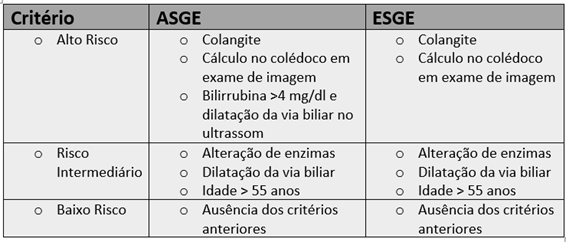
Pacientes com quadro de colangite ou cálculo visualizado no ultrassom de abdome ou em tomografia/RNM são considerados como de alto risco. A ASGE inclui um critério a mais, que é a presença de Bilirrubina total maior do que 4 mg/dl associada à dilatação da via biliar como critério de alto risco. Estes pacientes podem ser submetidos a CPRE direto, antes da colecistectomia e sem a necessidade de investigação adicional.
Portadores de alteração de enzimas hepáticas (TGO, TGP, FA, GGT) ou dilatação da via biliar no ultrassom (>6 mm com vesícula in situ) são considerados de risco intermediário. Novamente a ASGE tem um critério a mais que é a idade maior do que 55 anos. O racional para este critério é que pacientes com mais de 55 anos tem uma maior incidência de coledocolitíase não associada à alteração de enzimas hepáticas. Os dois guidelines sugerem que os pacientes com critérios intermediários sejam submetidos à colangiorressonância ou ecoendoscopia para avaliar a presença de coledocolitíase antes da colecistectomia. Na impossibilidade de realizar estes exames, a colangiografia intraoperatória é indicada como alternativa.
Já os pacientes que não apresentam nenhum destes critérios são considerados como de baixo risco e podem realizar a colecistectomia direto, com ou sem colangiografia intraoperatória. Nos critérios de 2010 a pancreatite aguda era um critério de risco intermediário. Porém, estudos confirmaram que a incidência de coledocolitíase residual após um episódio de pancreatite leve varia de apenas 10 a 30%. Devido a isso ela deixou de ser critério de risco. Os pacientes após a resolução da pancreatite aguda devem ser avaliados de acordo com os critérios acima. Se não apresentares critérios de alto
Qual dos critérios é melhor? ASGE ou ESGE
Um estudo incluindo 1042 pacientes comparou os critérios da ASGE e da ESGE para a avaliação da presença de coledocolitíase. Os resultados mostraram que os dois critérios são válidos e muito bons, com especificidade de 96,87% para os da ASGE e 98,24% para os da ESGE.
Comparando os dois o da ESGE é ligeiramente mais específico, com os critérios da ASGE apresentando um número discretamente maior de pacientes falso positivos.
Conclusão
A avaliação da probabilidade pré-teste em pacientes com suspeita de coledocolitíase é essencial. Esta avaliação pode identificar os pacientes que irão se beneficiar da realização da CPRE direto e também os pacientes necessitam uma avaliação mais aprofundada, evitando a realização de procedimentos desnecessários.
Referências
Manes G, Paspatis G, Aabakken L et al. Endoscopic management of common bile duct stones: European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy 2019; 51: 472–491
Maple JT, Ben-Menachem T. ASGE Standards of Practice Committee. et al. The role of endoscopy in the evaluation of suspected choledocholithiasis. Gastrointest Endosc 2010; 71: 1–9
Buxbaum JL, Abbas Fehmi SM, Sultan S, Fishman DS, Qumseya BJ, Cortessis VK, Schilperoort H, Kysh L, Matsuoka L, Yachimski P, Agrawal D, Gurudu SR, Jamil LH, Jue TL, Khashab MA, Law JK, Lee JK, Naveed M, Sawhney MS, Thosani N, Yang J, Wani SB. ASGE guideline on the role of endoscopy in the evaluation and management of choledocholithiasis. Gastrointest Endosc. 2019 Jun;89(6):1075-1105.e15.
Jagtap N, Hs Y, Tandan M, Basha J, Chavan R, Nabi Z, Kalapala R, Reddy PM, Ramchandani M, Gupta R, Lakhtakia S, Darishetty S, Rao GV, Reddy DN. Clinical utility of ESGE and ASGE guidelines for prediction of suspected choledocholithiasis in patients undergoing cholecystectomy. Endoscopy. 2020 Jul;52(7):569-573.
Como citar este artigo:
Orso, IRB. Como identificar e manejar os pacientes com suspeita de coledocolitíase? Gastropedia; 2022. Disponível em: https://gastropedia.pub/pt/cirurgia/hepatopancreatobiliar/como-identificar-e-manejar-os-pacientes-com-suspeita-de-coledocolitiase/