Câncer Gástrico Difuso Hereditário
No artigo anterior discutiu-se o câncer gástrico (CG) hereditário associado a presença de síndromes que ocasionam quadro de polipose digestiva.1 Nesse artigo será apresentado o Câncer Gástrico Difuso Hereditário (CGDH), a síndrome hereditária mais relevante sem associação com polipose do trato gastrointestinal.
1. CG Difuso Hereditário
O Câncer Gástrico Difuso Hereditário é uma síndrome caracterizada por uma alta prevalência de câncer gástrico difuso e carcinoma lobular de mama. Inicialmente descrita em uma família Maori da Nova Zelândia em 1998, estima-se atualmente que o CGDH tenha uma incidência populacional de aproximadamente 5-10/100.000 nascimentos.
A maioria dos casos confirmados de CGDH é causada por mutações germinativas no gene supressor de tumor CDH1. O CDH1 codifica a E-caderina, uma proteína transmembrana que está localizada nas junções celulares e tem funções de adesão, detecção de tensão e transdução de sinal através da membrana celular. A mutação do CDH1 com posterior alteração da E-caderina ocasiona a perda de coesão celular ocasionando o aspecto clássico de um carcinoma com células pouco coesas ou com células em anel de sinete. Mutações em um segundo gene também relacionado com a adesão celular, a alfa-catenina (CTNNA1), também são encontradas em uma pequena parcela de casos de CGDH.
2. Critérios Diagnósticos
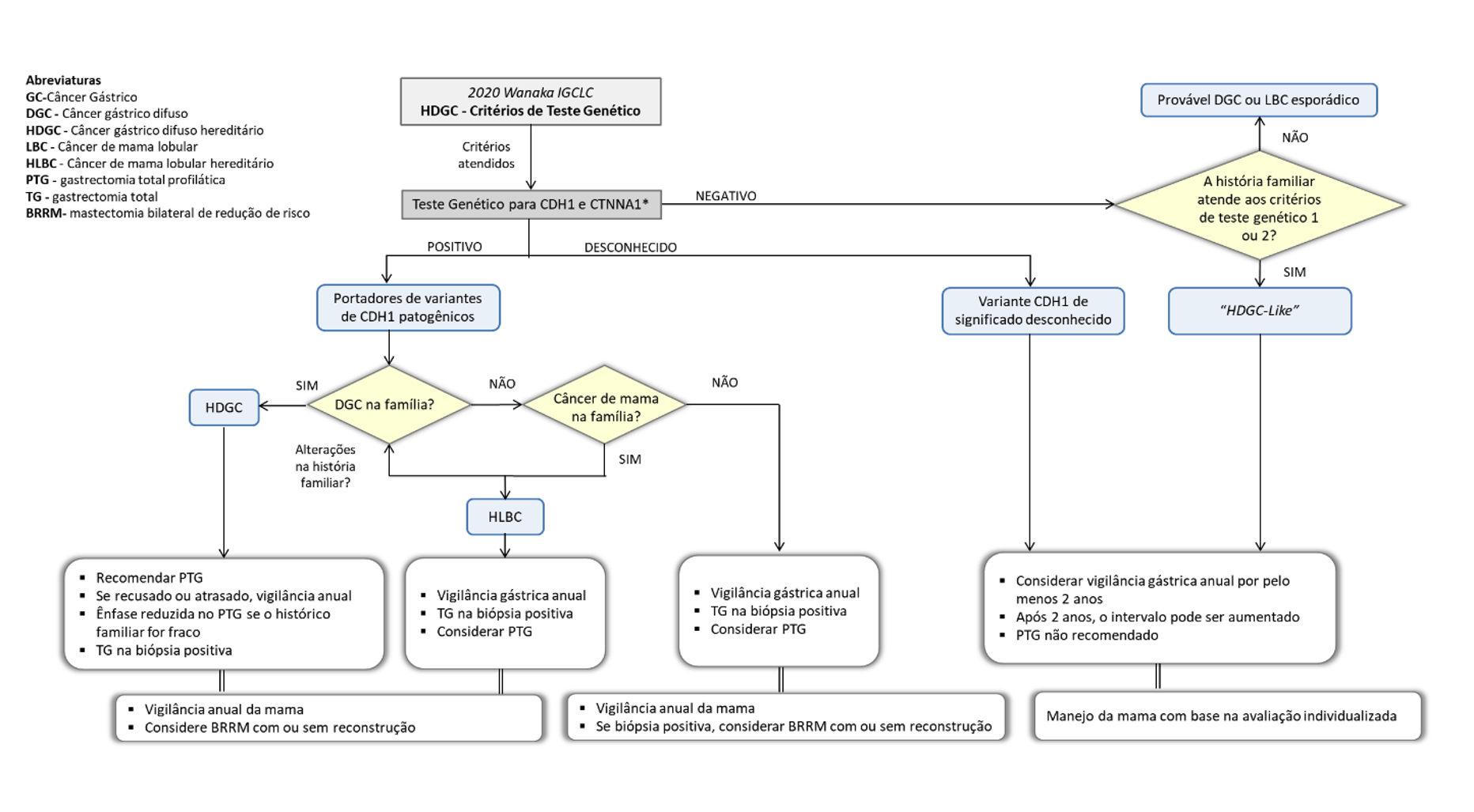
Os critérios de suspeita clínica tanto do indivíduo quando da família foram relaxados principalmente por meio de mudanças nas restrições de idade nas diretrizes de 2020 (Figura 1).2 A ampliação da indicação do teste genético deve equilibrar os custos relacionados à saúde, a aceitação do público e a carga psicológica imposta à população testada contra o benefício de identificar mais indivíduos assintomáticos de alto risco. O fato de o custo do teste estar diminuindo progressivamente também contribuiu para sua maior indicação.

Por outro lado, a introdução generalizada de painéis de genes de câncer, identificou variantes inesperadas de CDH1 em indivíduos que não apresentam fenótipos sugestivos de CGDH, criando um desafio para pacientes e médicos. Dessa forma, a pesquisa indiscriminada não tem indicação em casos sem suspeita clínica da síndrome. As estimativas de penetrância do para ocorrência de CG das variantes patogênicas de CDH1 são influenciados pelos critérios clínicos usados para indicar o teste genético. Usando famílias que preencheram os critérios clínicos anteriores de 2010, mais restritos para indicação do teste genético, estimou-se o risco cumulativo de CG aos 80 anos em homens e mulheres portadores de 70% e 56%, respectivamente. No entanto, outro relato evidenciou que famílias que atenderam ao critério de CGDH de 2015, menos rigorosos, a penetrância para ocorrência de CG foi 42% para homens e 33% para mulheres. Dessa forma, o risco de CGDH varia entre as famílias e, portanto, a história familiar deve ser considerada ao estimar o risco de penetrância de cada indivíduo.
Em indivíduos que atendam aos critérios clínicos para realização de teste genético, o teste costuma ser oferecido a partir da idade legal de consentimento (18 anos no Brasil). Sempre que possível, o aconselhamento genético para CGDH deve incluir avaliação de um pedigree familiar de três gerações, histórico familiar de lábio leporino ou fissura palatina e confirmação histopatológica do tipo de CG e/ou mama.
O aconselhamento deve ser realizado por profissional capacitado com discussão abrangente e multidisciplinar em torno dos benefícios e riscos da cirurgia gástrica profilática e vigilância do câncer.
3. Gastrectomia Profilática
Quando indicada, a gastrectomia total profilática (GTP) é realizada no início idade adulta, geralmente entre 20 e 30 anos de idade. Dado o aumento dos riscos perioperatórios e recuperação prolongada com a idade, a GTP não é recomendada em pacientes com mais de 70 anos, a menos que haja circunstâncias atenuantes significativas. Antes da cirurgia, os pacientes devem realizar uma endoscopia para garantir que já não haja CG, o que exigiria a realização do estadiamento completo. O exame também identificar outra patologia coincidente, como o esôfago de Barrett e hérnia de hiato, que pode alterar a extensão da ressecção.
A extensão da gastrectomia deve ser total, com confirmação intraoperatória de mucosa escamosa esofágica na margem proximal e mucosa duodenal na margem distal. As metástases linfonodais perigástricas são extremamente incomuns em pacientes submetidos a GTP na ausência tumor gástrico na endoscopia inicial. Dessa forma, uma linfadenectomia D2 estendida deliberada não é necessária e geralmente é desencorajada para minimizar a morbidade pós-operatória. Para evitar o potencial de subestimar o raro evento de um paciente com um tumor gástrico T2 anteriormente não identificado, a realização da linfadenectomia D1 é suficiente.
4. Vigilância Endoscópica
A vigilância deve ser realizada em centros especializados familiarizados com CGDH.
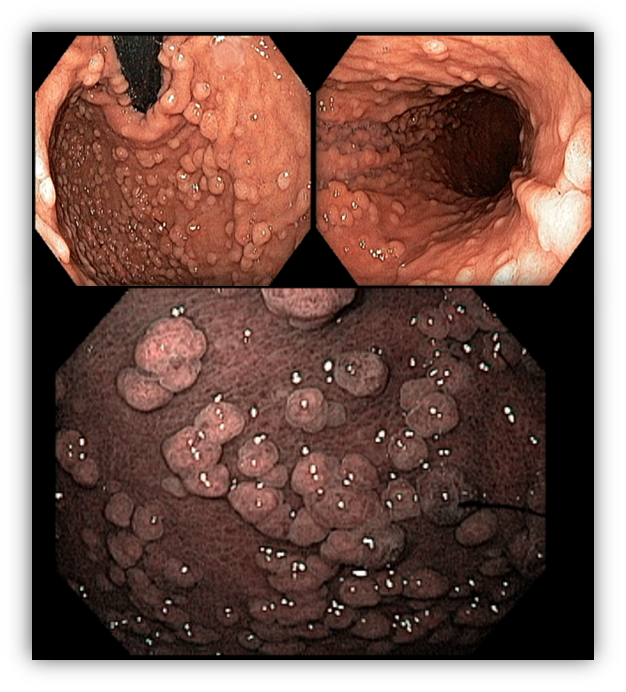
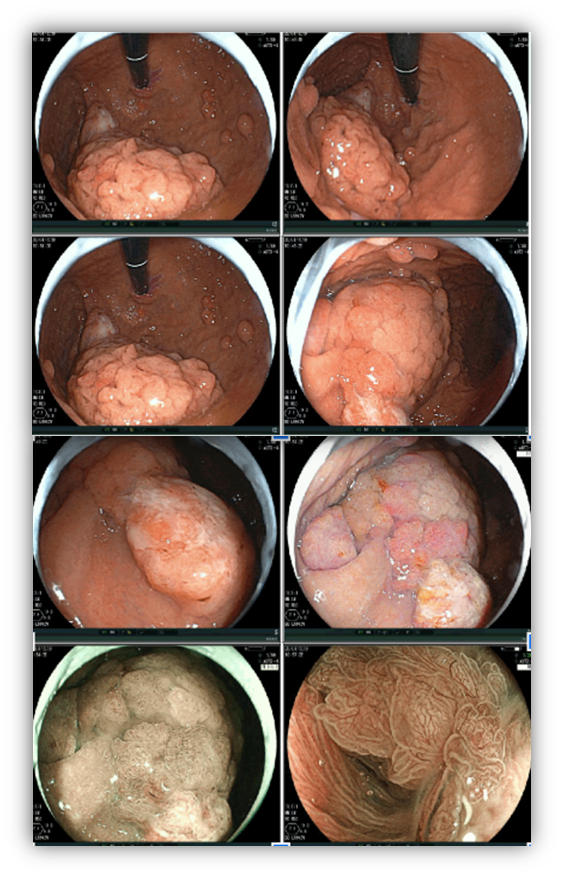
A chance a priori de ter pelo menos uma lesão compatível com carcinoma células em anel de sinete na amostra de gastrectomia total de um portador de mutação CDH1 é de 95%. Consequentemente, a relevância clínica de alguns casos de carcinoma superficiais (estágio T1a) em biópsias endoscópicas são questionáveis, especialmente porque esses focos de carcinoma em lesões superficiais podem exibir um comportamento muito indolente.
Portanto, o objetivo principal da vigilância não é apenas detectar um foco carcinoma superficial. Em pacientes que desejam adiar a cirurgia, outros objetivos da vigilância incluem: excluir lesões infiltrativas mais profundas, detectar grandes ou numerosas lesões T1a, pois esses pacientes provavelmente têm uma chance maior de desenvolver lesões em estágio T mais avançado, e avaliar a mudança histológica e aparência endoscópica que podem sinalizar comportamento mais maligno.
Dessa forma as endoscopias de vigilância devem incluir biópsias direcionadas e aleatórias. O número de biópsias aleatórias recomendadas é 28-30 (três-cinco na cárdia, cinco no fundo, dez no corpo, cinco na zona de transição corpo-antro e cinco no antro). Recomenda-se que as linguetas de tecido gástrico no esôfago sejam registradas, inspecionadas e biopsiadas. Todos os pacientes sob vigilância devem ser totalmente informados sobre as limitações do exame.
Referências
- Kodama, MFKP e Simoes IBP. Câncer gástrico hereditário. Gastropedia 2022. Disponível em: https://gastropedia.pub/pt/cirurgia/cancer-gastrico-hereditario
- Blair VR, McLeod M, Carneiro F, et al. Hereditary Diffuse Gastric Cancer: Updated Clinical Practice Guidelines. Lancet Oncol. 2020;21(8):e386-397.
Como citar este arquivo
Ramos, MFKP. Câncer Gástrico Difuso Hereditário. Gastropedia 2022. Disponível em: https://gastropedia.pub/pt/cirurgia/esofago-estomago-duodeno/cancer-gastrico-difuso-hereditario/