
El Síndrome de Gilbert (SG) es un trastorno hepático del metabolismo de la bilirrubina con reducción en la glucuronidación de la bilirrubina y consecuente hiperbilirrubinemia indirecta (no conjugada).
Es una condición común (3-10% de la población), con reducción en la actividad de la UGT1A1 en 25-40%. Las mutaciones ocurren en la secuencia de la región promotora (TATA box) del gen UGT1A1, que tiene la función de controlar los niveles de la proteína normal producida. De esta manera, en el SG, la proteína producida es estructuralmente normal, pero en menor cantidad.
| Gen UGT1A1 Promueve la producción de la enzima bilirrubina-UGT, responsable de la conjugación de bilirrubina. Por lo tanto, las mutaciones en la UGT1A1 generan la producción de una proteína anormal, con pérdida completa o niveles menores de actividad de la bilirrubina-UGT. |
Presentación clínico-laboratorial
Clínicamente, los pacientes suelen ser asintomáticos e identificar elevaciones en los niveles de bilirrubinas totales con predominio de bilirrubina indirecta (<4-5mg/dL), de forma incidental, o pueden presentar cuadros intermitentes de ictericia, en especial, desencadenados por gatillos como ejercicio físico intenso, baja ingesta calórica/ayuno, período menstrual, deshidratación e infecciones.
Laboratorialmente, no hay elevación de enzimas hepáticas o alteraciones en los demás exámenes de función hepática (tiempo de protrombina y albúmina), además de no haber indicios de hemólisis o enfermedad estructural del hígado.
Diagnóstico diferencial
Los trastornos en la captación hepática, almacenamiento, conjugación y excreción pueden causar hiperbilirrubinemia. Entre las causas hereditarias de hiperbilirrubinemia indirecta, con exámenes normales de función hepática y sin alteración de la histología hepática, además del SG, se realiza el diagnóstico diferencial con:
- Síndrome de Crigler-Najar tipo I: condición muy rara con herencia autosómica recesiva que se manifiesta poco después del nacimiento. Debido a la ausencia de actividad hepática UGT1A1, se produce ictericia grave (20-45mg/dL o más) y riesgo de daño neurológico y muerte por kernicterus (encefalopatía bilirrubínica) en los primeros días después del nacimiento. El tratamiento temprano para la reducción de los niveles de bilirrubina indirecta en la sangre es la fototerapia, debiéndose considerar la realización de trasplante hepático como única terapia curativa.
- Síndrome de Crigler-Najar tipo II: condición rara con herencia autosómica recesiva. Hay actividad hepática UGT1A1 de 10% o menos, con ictericia crónica (6-20mg/dL) y evolución potencialmente benigna. El tratamiento con fenobarbital propicia la reducción de alrededor del 25-30% de los niveles de bilirrubina indirecta por la inducción de la actividad de la UGT1A1 residual.
Investigación diagnóstica
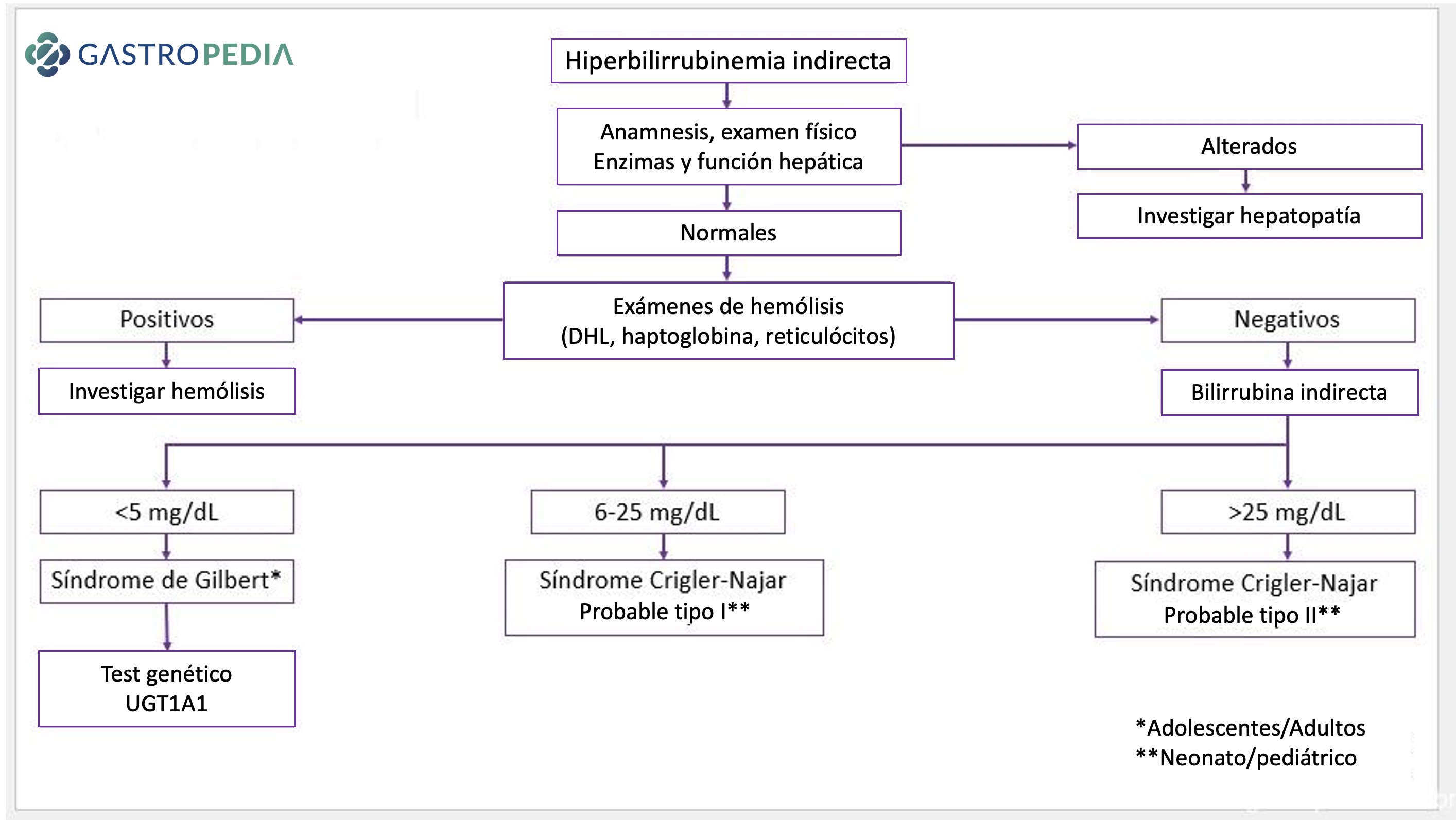
Identificada la hiperbilirrubinemia indirecta, se recomienda anamnesis y examen físico detallados, dosificación sérica de enzimas hepáticas (TGO, TGP, fosfatasa alcalina y GGT) y función hepática (tiempo de protrombina y albúmina).
Si hay alteraciones en esta primera etapa de evaluación, se dirige la investigación a la evaluación de hepatopatías, siendo prudente complementar con examen de imagen/ecografía de abdomen superior y demás exámenes laboratoriales específicos.
Si no se identifican alteraciones en la primera etapa de evaluación, es obligatorio descartar hemólisis con la dosificación de DHL, haptoglobina y reticulocitos.
En adolescentes o adultos, en ausencia de hemólisis y niveles de bilirrubina indirecta <5mg/dL, se presume el diagnóstico de síndrome de Gilbert. La confirmación se hace por la prueba genética para detectar mutaciones en el gen UGT1A1/TATA box.
Diagnóstico Genético
Ante la posibilidad de reacciones adversas a algunos medicamentos metabolizados por la UGT1A1, como el irinotecano y el atazanavir, se recomienda considerar la confirmación del SG mediante la investigación de la mutación UGT1A1 por el método de PCR en tiempo real (Imagen 1).
Cuando en homocigosis, no hay necesidad de rastreo adicional, sin embargo, si el paciente posee solo un alelo de la mutación UGT1A1 o ambos alelos son normales, se debe investigar las mutaciones G71R y Y486D, que también se asocian con el SG. Imagen 1. Resultado de la prueba genética para el Síndrome de Gilbert con homocigosis del alelo 28 en el gen UGT1A1 (en portugués).
Imagen 1. Resultado de la prueba genética para el Síndrome de Gilbert con homocigosis del alelo 28 en el gen UGT1A1 (en portugués).
Tratamiento
Por ser una condición benigna, el tratamiento es conservador solo con observación. El pronóstico de los pacientes con SG es excelente y no requiere tratamiento específico.
Referencias
- Thoguluva Chandrasekar V, Faust TW, John S. Síndrome de Gilbert. 2023 Feb 6. En: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 29262099.
- Singh A, Koritala T, Jialal I. Hiperbilirrubinemia no conjugada. 2023 Feb 20. En: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 31747203.
- King D, Armstrong MJ. Resumen del síndrome de Gilbert. Drug Ther Bull. 2019 Feb;57(2):27-31. doi: 10.1136/dtb.2018.000028. PMID: 30709860.
- Wagner KH, Shiels RG, Lang CA, Seyed Khoei N, Bulmer AC. Criterios diagnósticos y contribuyentes al síndrome de Gilbert. Crit Rev Clin Lab Sci. 2018 Mar;55(2):129-139. doi: 10.1080/10408363.2018.1428526. Epub 2018 Feb 1. PMID: 29390925.
- Rodrigues C, Vieira E, Santos R, de Carvalho J, Santos-Silva A, Costa E, Bronze-da-Rocha E. Impacto de las variantes del gen UGT1A1 en los niveles totales de bilirrubina en pacientes con síndrome de Gilbert y en sujetos sanos. Blood Cells Mol Dis. 2012 Mar 15;48(3):166-72. doi: 10.1016/j.bcmd.2012.01.004. Epub 2012 Feb 9. PMID: 22325916.
Cómo citar este artículo
Oti KST; Gamarra ACQ. Síndrome de Gilbert: ¿qué necesitamos saber? Gastropedia 2024, vol. 2. Disponible en: https://gastropedia.pub/es/gastroenterologia/sindrome-de-gilbert-que-necesitamos-saber
Gastroenterologia Clínica pelo Hospital das Clínicas da FMRP-USP. Hepatologia pelo Hospital das Clínicas da USP HCFMUSP. Doutorado em Ciências em Gastroenterologia HCFMUSP. Aperfeiçoamento de Elastografia Hepática Transitória HCFMUSP. Médica colaboradora do Ambulatório de NASH HCFMUSP. Membro FBG e SBH

Gastroenterología Pediátrica en el Hospital de Clínicas de la FMB-UNESP
Máster en Ciencias por la FMB-UNESP
Pediatra en el Hospital de Clínicas de la FMB-UNESP
Miembro Titular de la Sociedade Brasileira de Pediatria (SBP)
Miembro de la Sociedad Latinoamericana de Gastroenterología Pediátrica, Hepatología y Nutrición (LASPGHAN)

{kind=link}