Cistoadenoma Seroso de Páncreas
Los quistes pancreáticos son, en la mayoría de los casos, hallazgos incidentales de exámenes de imagen.
Se estima que alrededor del 3-14% de las personas sometidas a exámenes abdominales tienen como hallazgo alguna lesión quística pancreática. En estudios de necropsia, este hallazgo puede llegar al 24%. Se observa un claro aumento de prevalencia en grupos de edad mayores.
Las lesiones quísticas pueden dividirse en:
- quistes benignos: pseudquistes, quistes simples, cistoadenomas serosos
- quistes malignos: cistoadenocarcinomas, tumores neuroendocrinos quísticos, neoplasia sólido-quística pseudopapilar
- quistes con potencial de malignización: IPMNs y cistoadenomas mucinosos
En este artículo hablaremos un poco sobre el cistoadenoma seroso.
CISTOADENOMA SEROSO (SCA)
El cistoadenoma seroso es una lesión que afecta más a mujeres que a hombres (2:1), en la 6ª o 7ª década de vida.
Es una lesión que no tiene preferencia por ninguna región pancreática, pudiendo afectar la cabeza, el cuerpo o la cola de la glándula.
Aspecto radiológico
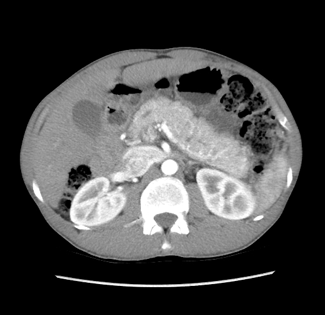
La característica más marcada del cistoadenoma seroso es el hallazgo de una lesión poliquística, con septos fibrosos entre ellos, formando un aspecto microquístico (70% de los SCA). En alrededor del 20-30% de los casos, los septos convergen hacia el centro de la lesión, formando una cicatriz central fibrosa (señal más típica del SCA). En el 20% de los casos observamos un aspecto de panal de abeja, con múltiples microquistes y septos finos entre ellos.


Figura 1: Cistoadenoma Seroso de cabeza de páncreas – lesión lobular con septos convergiendo hacia la ubicación central de la lesión. (archivo personal)
En alrededor de solo el 10% de los casos, los SCA pueden ser oligocísticos, lo que hace que el diagnóstico radiológico sea más desafiante. En estos casos, a menudo se necesitan otros exámenes para confirmar el diagnóstico, como la Ecoendoscopia con punción y análisis del fluido intracístico.
Características del fluido
La característica citológica del cistoadenoma seroso son células cúbicas, con citoplasma rico en glucógeno, aunque la sensibilidad para la citología con PAAF es muy baja.
El análisis bioquímico del líquido puede ayudar en los casos de diagnóstico incierto. La característica del SCA es tener el Antígeno Carcino-Embrionario (CEA) por debajo de 192 ng/ml, lo que se asocia a lesiones no mucinosas. Además, al no haber comunicación con los conductos pancreáticos, la amilasa en el líquido intracístico es baja.
Más recientemente, con el avance de la endoscopia confocal, es posible visualizar el patrón de vascularización (en los SCA, es subepitelial – precisión del 87%) y permite biopsias del epitelio del quiste. Este procedimiento se realiza aún en pocos centros, y aunque mejora la precisión del diagnóstico, conlleva mayores riesgos de efectos adversos (pancreatitis aguda y hemorragia intracisto).
Pronóstico
El pronóstico de los SCA es excelente, con menos del 1% de mortalidad. Pocos casos en la literatura han evolucionado hacia la malignidad, y no hay un acuerdo sobre la periodicidad del seguimiento. Para muchos autores, se trata de una lesión benigna.
Aunque es una lesión con baja probabilidad de transformación maligna, existe la posibilidad de crecimiento de la lesión en hasta el 40% de los SCAs.
La última recomendación del grupo europeo es realizar un nuevo examen de imagen en 1 año, y posteriormente, solo si hay síntomas (dolor abdominal, ictericia o náuseas y vómitos).
Cómo citar este archivo
Marzinotto M., CISTOADENOMA SEROSO DE PANCREAS. Gastropedia, 2022. Disponible en: https://gastropedia/gastroenterologia/pancreas/cistoadenoma-seroso-de-pancreas
Referencias
- Sakorafas, GH et al. Revisión de las neoplasias quísticas primarias del páncreas. Parte I: Neoplasias quísticas serosas. Oncología Quirúrgica, 2011
- Tirkes, T et al. Neoplasias quísticas del páncreas; hallazgos en la resonancia magnética con correlación patológica, quirúrgica y clínica. Imagen Abdominal, 2014
- Larson, A et al. Historia natural de los quistes pancreáticos. Dig Dis Sci, 2017